Clinical Trials Regulation (EU) No 536/2014
Introduction
This blog gives an overview of the Clinical Trials Regulation EU (No) 536/2014 (referred to as CTR); the CTR governs the procedures for conducting clinical trials in the EU and came in to force on 31st January 2022; it repeals the Clinical Trial Directive 2001/20/EC (CTD) which was previously applicable and can now be used by Sponsors for the following procedures:
An initial clinical trial authorisation application
Applications for substantial modifications of clinical trials
The supervision of clinical trials and
The application to add further Member State(s) to authorised clinical trials.
For the purposes of convenience, I have used ‘European Union (EU)’ to represent countries in both the EU and the European Economic Areas (EEA). As I have stated in a previous blog, all EU countries are part of the EEA, however, not all EEA countries are part of the EU.
A fairly broad overview of the objective of the CTR is given on the European Medicines Agency’s (EMA) website; in general the objectives of CTR are to:
Harmonise the processes for assessment and supervision of clinical trials throughout the EU. The intention is to make the EU an attractive region to conduct multi-national clinical trials and foster innovation and research.
Ensure a high standard of safety for all participants in EU clinical trials, and to
Increase the transparency of information and data obtained from clinical trials.
The business tool that enables the application of the CTR is the Clinical Trials Information System (CTIS); the CTIS is available for use by Sponsors, Member States, the European Commission and members of the public since 31st January 2022 when the CTR came into force.
An overview of the Clinical Trial Regulation (CTR)
The principles relating to the rights, safety, dignity, and well-being of subjects in a clinical trial, are embodied into the CTR.
The CTR allows for the scientific and ethical aspects of a clinical trial to be assessed at the same time, for multiple Member States, as a single application, if the Sponsor requires this.
This harmonised and streamlined approach reduces the administrative burden for Sponsors making the EU an attractive place to conduct clinical trials. It is particularly advantageous for rare or ultra-rare conditions where patient population from a larger pool of countries, (therefore several Member States), would likely be required. This can be progressed with one submission package and one application instead of the multiple submission packages and applications required for each Member State, as was the case with clinical trials conducted in accordance with the Member States’ national legislation transposed from the Clinical Trial Directive (2001/20/EC). For rare or ultra-rare conditions, more Member States can be added to an ongoing clinical trial, if patient recruitment is challenging; therefore the CTR is convenient for Sponsors by allowing this pragmatic approach.
Sponsors can add Member States on a case by case basis in an expedited manner (compared to the CTD) only after a positive decision for the authorisation of a clinical trial has been issued. The additional Member State is not required conduct a full scientific assessment, since this has already been completed during the initial clinical trial authorisation application. The additional Member State will be expected to review the already approved the scientific assessment report generated in the initial application (discussed in more detail see ‘Part I assessment’ below), and they can still raise comments on, or disagree with, this scientific assessment. They will also conduct their own ethical review for compliance with their national requirements.
This harmonised and streamlined approach is also applied to subsequent substantial modifications, (see below), and to updates of the CTIS database documents, required for the supervision of clinical trials.
Notwithstanding all of the above, there still remains hurdles for clinical trials in the EU. One example is the requirement for authorisation of the use of ‘Genetically Modified Organisms’, specifically, gene therapy medicinal products (GTMP) in clinical trials; these authorisations are currently managed at the Member State level and in accordance with their national legislation.
The CTR can be referred to directly by Sponsors; the necessity for country specific knowledge or expertise is limited to the ethical aspects; these ethical aspects are also defined in the CTR; additionally, the EMA has provided harmonised templates for utilisation and guidance documents. The templates and guidelines are available on the European Commission website for clinical trials: EudraLex Volume 10.
Ultimately the benefits expected are: to make the EU a more attractive region for conducting multi-national clinical trials; to encourage the development of innovative technology allowing for marketing authorisation of these products in the EU and improving the access of medicines, particularly for conditions that are debilitating, life-threatening and where no therapeutic treatment is currently available.
The transition period of 3 years for all clinical trials to be comply with the CTR started as of 31st January 2022. Next year, all new applications must comply with CTR, and all trials must be transferred to the CTR and managed on the CTIS by the end of 2025.
The CTR adopts a risk-based approach, where the risk to the subject(s) in the clinical trial is prioritised for assessment at an EU-level; at the same time this approach reduces unnecessary EU supervision for low-risk trials.
Aspects addressed in the CTR are:
Clinical trials and clinical studies (Chapter I, Article 2).
The concept of co-sponsorship (Chapter XI, Article 72).
Clarification and provisions for informed consent (Chapter V, Articles 28 to 35).
Approaches for clinical trials conducted outside of the EU that are referred to in a clinical trial application within the EU. These trials have to comply with the regulatory requirements and must be at least equivalent to what is applicable in the EU in terms of Good Clinical Practice (GCP) (Chapter XIII).
Requirement for a summary report of the clinical trial for layperson and the information contained therein. (Annex V of the CTR).
Safety Reporting (Chapter VII).
The labelling for the IMP, Auxiliary Medicinal Products and authorised investigational medicinal products; CTR defines what can be excluded with the use of a centralised electronic randomisation system and the use of a centralised information system, provided that the safety of the subject and the reliability and robustness of the data are not compromised. (This information is contained in Annex VI of the CTR).
Recruitment of subject into a trial in the MSC must occur within 2 years of trial authorisation otherwise the trial lapses and a reapplication would be required.
And finally, a word on the CTIS; this is a validated electronic system and the data contained within it could potentially be used for other assessment purposes (such as Health Technology Assessments). Moving forward, it remains to be seen how or if data contained in the CTIS will be incorporated into the EU Data Quality Framework.
Clinical trials
The CTR provides more clarity on the definition of a clinical trial when compared to the information from the CTD; a clinical trial is a sub-category of a clinical study.
A clinical study is an investigation in humans intended to:
a) Discover or verify the clinical, pharmacological or other pharmacodynamic effects of one or more medicinal products; and
b) Identify any adverse reactions to one or more medicinal products; or
c) Study the absorption, distribution, metabolism and excretion of one or more medicinal products;
with the objective of ascertaining the safety and efficacy profiles of those medicinal products.
A clinical trial is a clinical study which fulfils any of the following conditions:
a) The assignment of the subject to a particular therapeutic strategy is decided in advance and does not fall within normal clinical practice of the Member State concerned.
b) The decision to prescribe the investigational medicinal product (IMP) is taken together with the decision to include the subject in the clinical study; or
c) Diagnostic or monitoring procedures in addition to normal clinical practice are applied to the subject.
The CTR defines a ‘Low-intervention clinical trial’ as a clinical trial that fulfils all of the following conditions:
a) The IMP, excluding placebos, are authorised
b) According to the protocol of the clinical trial:
i. The IMP are used in accordance with the terms of the marketing authorisation; or
ii. The use of the IMP is evidence based and supported by published scientific evidence on the safety and efficacy of those IMP in any of the Member States concerned; and
c) The additional diagnostic or monitoring procedures do not pose more than a minimal additional safety risk or burden to the subjects compared to normal clinical practice in any of the Member State concerned.
Clinical trials are within the scope of the CTR and should be managed at EU level. Specifically, these are low-interventional clinical trials and interventional clinical trials.
Non-interventional clinical trials are out of scope of the CTR and should be managed by Member States.
Also, out of scope of the CTR are:
Medical devices.
Trials without medicinal products (as defined by Directive 2001/83/EC).
Any study in humans for which the objective is not to investigate the effects of the IMP itself but, for example, to study the physiology of the human body as a result of taking the IMP. Here, the IMP is used as a tool with the aim to provoke a well characterized physiological response in humans. These studies should not address the diagnostic, prophylactic or therapeutic potential of the medicinal product nor its pharmacokinetic or pharmacodynamic profile. It is important to note that if the study switches and the objective evolves to become a study of the IMP itself, (and developers need to be aware and sensitive of this), then a clinical trial is applicable which should be determined, assessed and managed at the EU level.
Interventional clinical trials and low-intervention clinical trials, managed at the EU-level, are always confirmed on validation as compliant with the CTR (or not); therefore, it really is important for the Sponsor to clearly state and understand the objective of the clinical trial as eluded to above.
Clinical Trial Information System (CTIS)
The Clinical Trials Information System (CTIS) is the single-entry point for clinical trials authorisation and supervision in the EU. The CTIS consists of the EU Portal for communication between Member States and the Sponsor, a workspace for assessment, and a public website (or database) containing information on all the registered clinical trials for public view, (this includes clinical study report suitable for the layperson). The CTIS is the business tool that enables the application of the CTR.
The CTIS interfaces with the EudraVigilance website; safety reports still have to be submitted via the EudraVigilance website; however, the necessity to submit safety to reports to each Member State is removed, simplifying safety reporting of clinical trials. The CTIS also interfaces with the Organisation Management Services (OMS) database; the OMS database is where validated information (e.g. names and addresses and contact details) on Sponsors and/or manufacturers is located; the CTIS therefore provides a reservoir of information on registered clinical trials in the EU.
In addition, as previously stated, the CTIS is used for the supervision of Clinical Trials in accordance with Article 81.9 of the CTR. All the latest versions of critical documents (protocols, Investigators Brochure (IB) and Investigational Medicinal Product Dossier (IMPD)) are retained here for supervision.
Since the application on 31st January 2022, clinical trials have been conducted using the CTR and as of this date, I have found 79 clinical trial applications at various stages of assessment and approval on the CTIS database.
The search tools on the CTIS are intuitive and the database provides a lot of information in a suitable format for both members of public and the pharmaceutical industry worldwide. This is probably due to the fact that the CTIS was user tested by members of the public, academia and industry representatives during its development and construction. It can be used by anyone worldwide to search for clinical trials conducted in the EU; all that is required is a suitable device and internet access.
For consideration: the CTIS contains validated data in an electronically secure system; how this data is used could be further, in terms of secondary use, could be further defined in the forthcoming Data Quality Framework.
Assessment of Clinical Trial Applications (Articles 5 - 8)
CTR enables the scientific and ethical assessments to be combined during a single procedure for one or more Member States. The assessment procedure consists of:
1. The scientific aspects of a trial (Part I Assessment) led by the reporting Member State (RMS), and
2. The ethical aspects of a trial (Part II Assessment) conducted by each concerned Member State (MSC).
A minor point of clarity, the RMS is also a MSC and also has to conduct it’s ethical review as well leading and drafting the Part I Assessment review documents.
At the end of the procedure, a single decision from each EU Member State is communicated via the EU Portal to the Sponsor and a single European Commission decision on the trial is recorded.
Validation
The validation consists of two components:
Confirmation of the RMS, and the
Technical validation of the application (to confirm if the application is within the scope of the CTR and if the dossier provided is complete).
Confirmation of the RMS
The Sponsor is responsible for the selection of the RMS. It is during the validation period (which is 10 days) that confirmation of the acting RMS must occurs. The Sponsor selected RMS has 3 days to notify the other MSCs if it is unwilling to be the RMS and the RMS must be agreed between all the MSC and communicated to the Sponsor within 6 days.
Technical validation of the application
The validation of the dossier itself must occur within 10 days from the submission of the application dossier. The RMS validates the application taking into account the considerations expressed by the other MSC (by day 7). The following are addressed and notified to the sponsor:
a) Whether the clinical trial applied for falls within the scope of the CTR (i.e. interventional clinical trial, low interventional clinical trial)
b) Whether the application dossier is complete in accordance with Annex I of the CTR
The Annex I list of documents required for a clinical trial application are:
Cover letter
EU Application Form
Protocol
Investigator’s Brochure / Summary of Product Characteristic
Documentation relating to compliance with Good Manufacturing Practice (GMP) for the IMP (Manufacturing and Importation Authorisation licence)
Investigational Medicinal Product Dossier (IMPD) / Simplified IMPD (including Placebos and Auxiliary Medicinal Products)
Scientific advice and Paediatric Investigation Plan (PIP)
Content of the labelling of the IMPs
Recruitment arrangements (information per MSC)
Subject Information, Informed Consent Form and Informed Consent Procedures
Suitability of the Investigator (information per MSC)
Suitability of the facilities (information per MSC)
Proof of insurance cover or indemnification
Financial and other arrangements (information per MSC)
Proof of payment of fee (information per MSC) and
Proof that the data will be processed in compliance with Union law on data protection
If any documents are missing, the RMS must notify the Sponsor via the EU portal and set a maximum of 10 days for the sponsor to comment or provide the additional information through the EU portal.
The RMS must confirm within 5 days of receipt of the comments or additional documents if the application is complete and complies with the requirements stated above, via the EU portal. The date that this confirmation occurs is the date of validation. If the RMS does not notify the Sponsor within this time, the tacit approval of validation is applied and the Sponsor can assume that the application is valid.
Part I Assessment
The Part I Assessment is led by the RMS. It is the scientific assessment and consists of the following phases:
The initial assessment phase: this is performed by the RMS and involves the development of a draft Part I assessment report; it should be concluded within 26 days of the validation date;
The coordinated review phase: this is performed within 12 days from the end of the initial assessment phase and involves all the MSCs; the draft assessment report is circulated, reviewed and commented on by the MSC; and
The consolidation phase performed by the RMS within 7 days from the end of the coordinated review phase: the considerations relevant to the application are addressed and recorded by the RMS.
In total Part I Assessment must be completed within 45 days from the validation date and involves the review of the following:
Low interventional trial
Benefit/risk profile
Quality (Investigational Medicinal Product Dossier) / Auxiliary Medicinal Product Dossier (if applicable)
Labelling
Investigator’s Brochure / Summary of Product Characteristics (SmPC)
Paediatric Investigation Plan (if applicable)
Protocol
Assessment report
If additional information is required for completion or authorisation, then this can be requested by the RMS via the EU portal.
In this scenario, the Sponsor has a maximum of 12 days to respond and provide the information. If the information is not provided within those 12 days (or the timeline stated by the RMS) then the application is deemed to have lapsed in all the MSCs and the Sponsor will have to make a reapplication to the MSCs.
Following receipt of the additional information the following steps occur:
Coordinated review of the additional information by all the MSCs – this takes 12 days
Consolidation phase by RMS – this takes 7 days.
In total, 31 days is added to the procedure for the request and review of additional information.
It should be noted that these timelines can be accelerated if required, particularly for urgent applications giving the option to expedite authorisation for access to IMPs.
Part II Assessment
The Part II assessment is performed by each MSC and relates to the ethical and national requirements for each of the Member State. The MSC decides how to involve the national competent authority and the Ethics Committee in the assessment to reach a decision. The role of the Ethics Committee is governed by each MSC; the Ethics Committee should include a layperson or a relevant patient group during their review.
The Part II Assessment must be completed within 45 days of the validation date; it includes:
Informed consent/Subject information
Recruitment
Suitability of trial centres and investigators
Data protection
Damage/financial compensation and
Biobanking.
It should be noted that the CTR does allow for the possibility for MSC to disagree with Part I assessment, but these are limited to the conditions of:
The subject would receive an inferior treatment than normal clinical practice in that MSC.
Infringement of their national law (e.g. clinical trial of a medicinal product that is forbidden in that MSC).
Specific concerns relating to the reliability and robustness of the data.
Also, the MSC can refuse the application if their national ethics committee has issued a negative opinion for the trial in that MSC.
If additional information is required for completion or authorisation during the procedure, then this is communicated by the MSC to the RMS; the RMS then request this information from the Sponsor via the EU portal.
In this scenario, the Sponsor has a maximum of 12 days to respond and provide the information. If the information is not provided within those 12 days (or the timeline set by the RMS), then the application is deemed to have lapsed in the MSCs and the Sponsor will have to make a reapplication.
Following receipt of the additional information the MSC has up to 19 days to review the additional information and approve or reject the application.
In total, up to 31 days is added to the procedure for the request and review of additional information, and this Part II Assessment request for further information can be coordinated to occur at the same time as Part I Assessment requests for further information, thus harmonising the whole procedure.
Within 5 days of the procedure a decision must be issued by the European Commission on the clinical trial application.
The decision can be:
The clinical trial can be authorised
The clinical trial can be authorised subject to conditions
The application is refused
Staggered Applications (Article 11)
The option to separate and stagger the Part I and II assessments is also available to Sponsors if they require it; this means that for some MSCs Part I can be submitted alongside other MSCs in the procedure who have received a full dossier for assessment of Part I and Part II.
In these cases, the Part II submission documents are staggered for the relevant MSC to a later date. However, for this approach, Substantial Modifications impacting Part I and Part I and II combined, cannot be submitted where there is an ongoing Part I and Part II assessment in another MSC for an initial clinical trial application. In these situations, the last Member State completing the Part II assessment determines the timings for a Substantial Modification. The Sponsor has two years to submit the Part II documents for the authorisation and they need to confirm that no additional information impacting on the integrity of the Part I Assessment report is available for the clinical trial to be authorised.
Substantial Modifications
These are any change to any aspect of the clinical trial which is made after notification of a decision for a clinical trial, and which is likely to have a substantial impact on the safety or rights of the subjects or on the reliability and robustness of the data generated in the clinical trial.
Therefore a substantial modification in the Clinical Trials Regulation can be considered only after a decision on an initial application or an application for substantial modification or addition of a MSC is granted. Substantial modification cannot be assessed while any assessment for the above is in progress. This includes staggered applications where Part I assessment only have been completed and a substantial modification impacting on Part I is proposed (please see Question 3.6 in the document Regulation (EU) No 536/2014 Questions & Answers May 2022). Substantial modifications can only be assessed after a positive decision on the previously submitted application is issued or authorised by tacit approval. This makes sense since all of the MSCs need to contribute towards the assessment and if there are assessments that are ongoing - then this would make it impossible for some staggered MSCs to perform two assessments on the same clinical trial at the same time.
A point to note is that Part II only substantial modification to specific MSCs can be progressed in parallel - so this is allowed.
Substantial modifications can be affected by the timings of ongoing MSC assessments, so they have to be suitably managed. It may well be that in order to submit a substantial modification, application currently ongoing in a MSC (such as an ongoing Part II assessment) may need to be withdrawn. Careful planning and communication with the Sponsor’s regulatory affairs department and the EMA at the earliest stages of development is required.
Annex IV of the EMA’s Question and Answer document has a list of classification of changes, to aid the identification of substantial modifications.
Subsequent Addition of another Member State
A sponsor can submit an application to add another MSC if required. This is a 52 day procedure (however, it can be extended by an additional if there are validation issues (10 days) or a requirement Requests for Further Information (31 days)).
The procedure consists of the MSC assessing the Part I assessment report (from the previous initial assessment or a modified version due to a substantial modification) and their separate Part II assessment for national requirements. For Part I assessment the additional MSC can only disagree with the positive decision if:
The subject would receive an inferior treatment than normal clinical practice in that MSC.
Infringement of their national law.
Specific concerns relating to the reliability and robustness of the data.
The MSC can also refuse the application based on the opinion of their national Ethics Committee.
An additional 31 days is provided for the assessment if a request for further information is made. This is for the review of the additional data. In this scenario, the sponsor has 12 days to provide the additional information and the MSC has 19 days to review the information before providing an opinion.
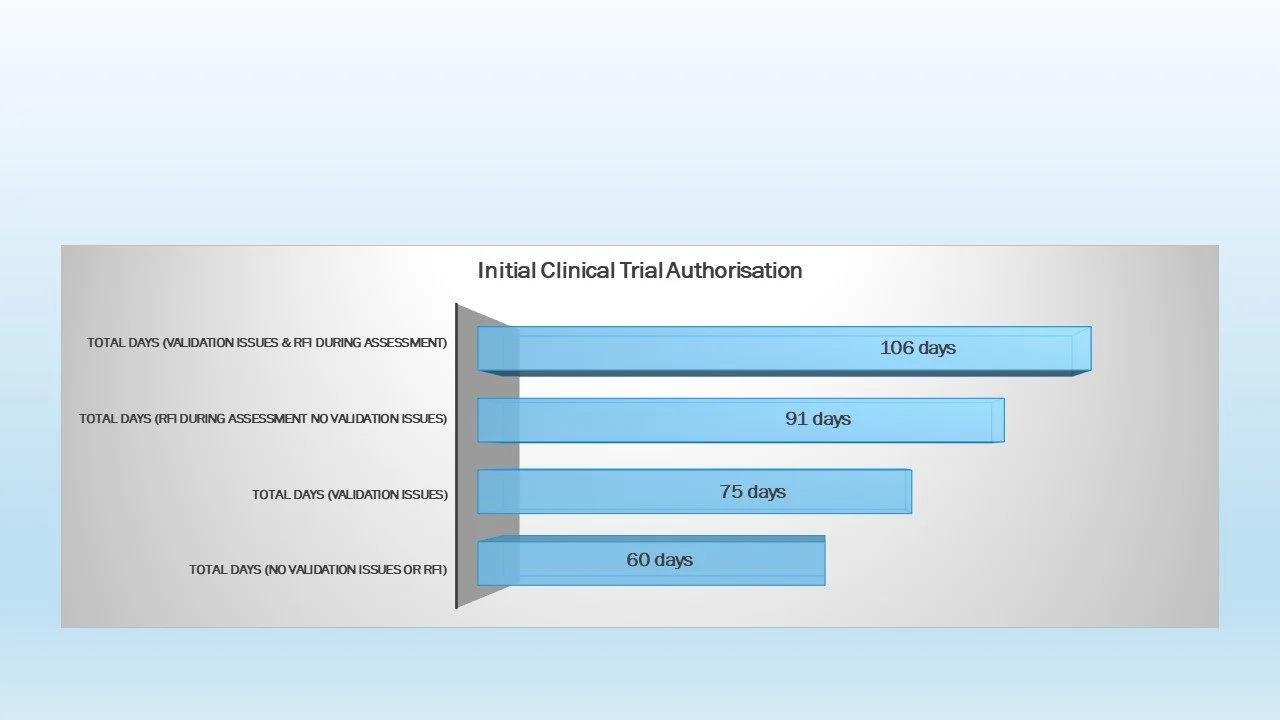
An illustration of the timelines…


Transparency
The clinical trial regulation has introduced major changes to the application and regulation of clinical trials in the EU. It has harmonised and streamlined the applications for ethical and scientific reviews. Efficiencies are achieved in regard to safety reporting, all of the clinical trial dossier documents are stored in the secure, validated CTIS database, and information on clinical trials should be made public, in a format that is understandable to the layperson.
The EMA is committed to transparency, EMA’s Policy 0070 relates to the access to data (ATD), example on the EMA’s website include, the information published for Marketing Authorisations (e.g., EPARs / Product information and Questions and Answers on the medicinal product for layperson); this was in response to the public demand, for the EMA, to publish information from which decisions were based.
For clinical data generated from clinical trials the same approach to transparency is adopted and this is detailed in the CTR. Most of the information submitted through the EU portal (particularly clinical study information) will be made public. Quality information, (Chemistry Manufacturing and Controls), is considered commercially confidential information (this includes the Investigational Medicinal Product Dossier, Regulatory Scientific advice, etc). Personal data is protected by Member State legislation and EU law and therefore also remains confidential.
The EMA promotes transparency in order to:
avoid duplication of clinical trials so that innovation and development of new medicines is encouraged
ensure public trust and confidence in the EMA’s scientific decision making process
enable public scrutiny of clinical trial data whilst protecting personal data and commercially confidential information.
Information on clinical trials is already published on the EU Clinical Trial Register. The CTR legislates that ‘clinical’ information must be made available in a format understandable to the layperson and it also legislates that one aspect of the CTIS functionality should be to function as a searchable database for public information on clinical trials in the EU. There are guidelines and templates available for Sponsors on how to author and format summaries of clinical trial results for the laypersons.
Justification by the Sponsor as to why any specific clinical information should not be made public will be required to redact information from clinical study reports. The EMA’s assessment policy for public access is such that it does not accept the redaction of information as commercially confidential if: a) the information is already available in the public domain; b) the information does not bear any innovative features; c) the information is common knowledge within the scientific literature / community; d) if the justification provided by the Sponsor is irrelevant to the text proposed for redaction and e) if the commercial harm is not explained or insufficiently explained.
My thoughts are that Sponsors should work closely with their legal teams early on in the product development stages in regard to identification of product sensitive information that could be included in the clinical study reports and consider how this information could be presented.
Another point to note, the EMA is already involved in international collaboration with other countries (Canada, United States of America and Japan), in regard to sharing of information and process relating to transparency.
Conclusion
This is the realisation of an ambitious project; there has been a few delays (!) and a long build up; the delays were due to technical and validation hiccups with the CTIS, it was important to get these technical issues resolved due to the nature and role of the CTIS for the regulation of clinical trials. In my opinion, this is a great achievement by the EMA, bearing in mind the changes they were undergoing before, during and after Brexit; the UK played a significant role in CTR and the CTIS. The roll out of the CTR once the CTIS was up and running earlier this year seems to have been successful.
This is a significant benefit for innovative medical technology, particularly for conditions that are debilitating and life-threatening; these are usually rare or ultra-rare diseases. By reducing the administrative burden for Sponsor when making application to multiple Member States, and increasing flexibility and options for Sponsors, the EMA is facilitating the development of innovative and/or new medicines as well as aiding in the understanding of the safety and efficacy profiles of authorised medicinal products. The harmonised authorisation process for clinical trials, the advantageous option to expedite the addition of Member States to clinical trials, and the ability to stagger applications for Part I and Part II assessments, illustrates that the CTR has built in flexibility for Sponsors to encourage the development of all modalities of medicinal products.
The challenges for Sponsors are to ensure that they submit high quality documents since there will not be a significant amount of time to generate new information during clock stop periods; this is repeatedly emphasised by the EMA. The time limitations for Sponsors to provide additional information might be particularly challenging for Advance Therapy Medicinal Products (ATMP); although an additional 50 days is added to the initial assessment phase for the RMS and the MSCs, the Sponsor has a limited time (12 days) to provide any additional requested information compared to CTD - so things have changed. Developers for ATMPs and other complex biological medicines could consider obtaining scientific advice before they submit their applications; in this case, the regulatory affairs department should be strengthened so as to ensure checking and approval of dossiers for clinical trial application. Sponsors will also be eager to foster good relationships with the EMA.
There could be potential issues around the selection of a RMS by Sponsors. I am thinking specifically of a scenario where the RMS selected is not acceptable to the other MSCs. Certain MSCs may have more exposure, expertise/experience with clinical trials than other MSCs; 6 days to resolve this seems quite ambitious to me, however, so far, it appears to be working.
Regarding the reporting of safety information; it is far less of a burden to report one set of safety data instead of the same data and information to several separate Member States; therefore, efficiency improvements in safety reporting, must (in my opinion) filter down to the safety of the subjects of clinical trial, therefore improving their well-being, and care - this is a good thing.
The transparency of clinical trial information to the EU and worldwide public has also been realised; the CTIS is fulfilling its role as a validated database by providing information to the layperson, and as I mentioned previously, all that is required to obtain this information is a device with access to the internet.
Clinical trials are not limited to big pharma, or small to medium sized pharmaceutical enterprises. Academia are also stakeholders and sometimes do not have the resources or the regulatory professionals and/or expertise to progress complex clinical trial applications. The EMA offers online training modules specifically to address some of these concerns.
As with any new process or procedure, there will be a learning curve and time required for the procedure to bed in for all stakeholders. The CTR is reviewed every 5 years as defined in the Regulation itself (Article 97). This review consists of an assessment of the impact that the Regulation has had on scientific and technological progress to ensure that the EU remains a competitive region for conducting clinical trials. The inevitably optimisation to the CTIS functionality as time progresses and more experience is gained and feedback is given, will ensure that the CTIS is a more efficient tool for the management of clinical trials.
The future beckons…
Best wishes,