Quality Risk Management
Introduction
Quality Risk Management (QRM) is a systematic approach designed to improve quality by identifying, analysing and evaluating potential risks and hazards associated with products manufactured, distributed and marketed in industry.
The QRM approach is also used in other industries such as food and biocides.
In the pharmaceutical industry, the QRM approach is integrated into the company’s Pharmaceutical Quality Systems (PQS), via established processes and/or procedures; these include work instructions, standard operating procedures (SOPs), and policy documents.
A PQS is a comprehensive model based on International Standards Organisation (ISO) quality concepts; the purpose is to make available good quality medicines, throughout all of the different stages of a medicinal product’s lifecycle. Quality medicines is attained by adherence to the regional standards of good practice (e.g. Good Manufacturing Practice, Good Distribution Practice, Good Laboratory Practice, Good Clinical Practice, Good Pharmacovigilance Practice etc.).
As previously stated, QRM is a systematic approach of looking at risk. Risk is defined as the combination of the probability of the occurrence of harm and the severity of that harm. A risk assessment is prospective exercise that identifies, defines and/or describes, through qualitative and/or quantitative measurements, a ‘hazard’ which is a potential source of harm. In pharmaceuticals, the aim of a risk assessment is to identify risks or hazards to prevent direct or indirect harm to patients, i.e. to promote and improve patient safety.
Discussion
A QRM requires the collaboration of different stakeholders / departments to contribute towards defining, assessing, analysing and making final risk-based decisions.
The perspectives of risks in terms of harm and severity, differ according to the concerned stakeholders, therefore it is important to obtain different perspectives and view points.
QRM utilises specific tools, (some are described below), in order to assess the risk and make the best case scenario decisions in accordance with the evidence available at the time; these decisions should be documented (even if they are informal low risk assessments and certainly for formal high risk assessment), in accordance with the processes in place. They could be reviewed or reassessed again at some later defined date or periodically whichever is more appropriate.
So, the purpose of the QRM is to provide a structured process to perform risk assessments; decisions must be based on scientific knowledge and reasoning, evidence or other verified information; the amount of effort and formality of the assessment should be commensurate with the level of the defined risk.
The objective of a risk assessments is to be able to make informed decisions based on the available data, information and perspectives at that time; these decisions could be reviewed and revised if, or when, more information becomes available, at a later date.
In order to make the best risk-based decisions, any assumptions, uncertainty, or gaps in knowledge should be clarified and made transparent to all the concerned stakeholders.
The steps for a QRM process could include the following:
Define the scope of the risk assessment.
Perform risk assessment - includes: Hazard Identification/Analysis/Evaluation
Document (formal or informal) the assessment (this could include a protocol with associated document references) and produce a report and/or an output; communicate report (and protocol) to all the concerned stakeholders.
Review of output by stakeholders.
Determine an agreed risk-based decision.
Implement the decision - this could include the agreed ‘Risk Control’ measures.
Perform a Risk Review.
It should be noted that the implementation of risk control measures could occur at any time, dependent upon the nature of the risk and the urgency for intervention; the department generating the data; and the potential for harm, hazard, damage or loss of product. Risk control measures and risk review should be applied by a case by case basis in accordance with the company’s PQS.
Risk Assessment:
This is a prospective exercise where risk or hazards are identified, analysed and evaluated. The identification of the risk/hazard usually occurs before an event (otherwise it is no longer a ‘risk’ and should be treated as an ‘issue’ although risk assessment tools and approaches are still useful). The ability to detect a risk should also be assessed if relevant. Absence of evidence of risk does not necessarily equate to the absence of the risk.
Any final documentation produced from the risk assessment should be retained in a controlled manner. The risk assessment documentation could be derived from an agreed template which could be considered as a backbone document; whenever a risk assessment is preformed the document (given a version number and considered a ‘living document’) is updated with the new information/evidence or practices. It could provide historical data so that future events can be assessed based upon documented experience (as opposed to relying on the experience of personnel who may or may not be employed at the manufacturing facility at the time). Risk assessment ‘living documents’ used in the pharmaceutical industry could be used for comparability purposes (such as manufacturing changes) and/or the information could be included in the pharmaceutical development sections of a dossier.
The SMARTER approach could be applied to risk assessments. This is as follows:
S - Specific - the hazard should be specifically identified and the assessment focussed on that particular hazard. This can be achieved by framing the risk as a question.
M - Measurable - the effect of the hazard should be either qualitatively or quantitatively defined in terms of severity of harm and probability of occurrence.
A - Assignable - the actions and owners of the actions should be specified.
R - Results - information and data generated through the risk assessment process.
T - Time-bound - a completion date should be specified and agreed.
E - Evaluation - perform a risk review of output by all stakeholders.
R - Record - decisions and actions should be documented for transparency and knowledge retention.
A diagram of an example of a QRM process, extracted from the ICH guidelines on QRM, is illustrated below:
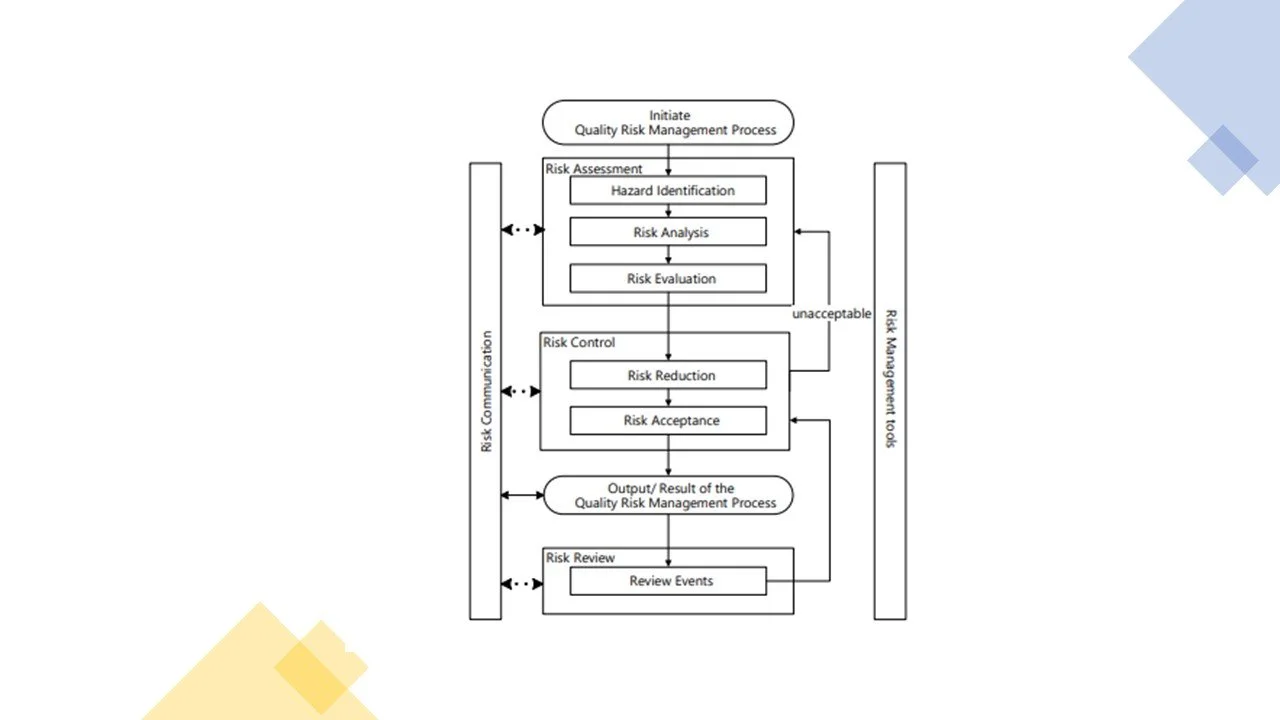
Formality of Risk Assessments and Risk Based Decisions
The formality of risk assessments is dependent upon the ‘hazard’ or risk under considerations; the impact of the hazard on product and the areas affected by the hazard or the stakeholders or department affected by the hazard. Risk assessments that are high in hazard and quality impact will require a more formal assessments and risk based decisions.
Hazards that are considered low in terms of impact to patient safety could require less formal assessments; they may however require formal risk based decisions if they impact on specific areas of the pharmaceutical business - such as sales and marketing. The emphasis of all risk-based decisions should be on patient safety.
The effort, formality and the procedures for risk assessments and risk based decisions should be determined on a case by case basis and the work instructions, SOPs and policies available within the company’s PQS, should be chosen dependent on the effort required due to the impact of the risk or hazard.
Generally, formal assessments involve more documentation following more standard processes (such as protocol preparation, report generation); assessments are undertaken using the company’s ‘Change Control Systems’ etc. Flexibility should be built into the processes and systems to allow for informal assessments where less documentation is required. Informal assessments would still fall under the company’s ‘Change Control System’ to enable the knowledge to be retained.
A more comprehensive guidance of the requirements for risk assessments and risk based decisions are given in the ICH Q9(R1) guidelines for Quality Risk Management. It should be noted that this guideline is currently under review.
Hazard / Risk Identification:
The first step of a risk assessment is to define the scope for a focussed identification of hazard; the aim is to ensure patient safety through science based decisions and it is important to keep this objective in mind for all discussions and decision making process. As previously eluded, identification of risk can depend upon perspective; one stakeholder’s view of risk, may differ to other stakeholders. As long as the objective is ‘patient safety’, and this takes precedence the most appropriate informed decisions can be made.
Risk Analysis:
This relates to a qualification or quantification assessment for the concerned risk. It should assess the likelihood of occurrence and the severity of harm using the tools described below and other techniques to qualify and quantify the risk. The risk analysis may involve carrying out testing (thus working to an approved defined protocol) to provide data to assess changes where risk are involved.
Risk Evaluation:
The risk analysis exercise involves the collection of data and information; this should be compared and/or discussed against a predefined set of criteria during the evaluation phase and the conclusions of which reported in the output.
Risk Control:
Following on from the risk assessment, the data yielded should provide information on the decisions for the next steps with respect to the concerned risk. Risk control measures include steps to control or reduce the risk, such as controls in place for raw materials and critical starting materials. The strategies for risk control involve the following:
Avoidance - a method to mitigate risk by not participating in activities that may negatively impact on quality.
Retention - this involves the accepting the risk.
Sharing - ownership of quality standards is accepted by partners and suppliers.
Reduction - taking activities to reduce the defined risk if the action itself does not introduce risk.
Communication:
It is important to ensure visibility and transparency of all risk assessments to all of the stakeholders; sometimes it is not immediately clear who needs to be involved during the risk assessment and this may need to be continually reviewed. Reviews could occur during during change control meetings, quality assurance meetings or other meeting with specified collaborators involved in the risk based decisions. An agreed distribution list should be part of the QRM and PQS procedures.
The output of the risk assessment - the report, should be clear and easily digestible, such that reviewers can quickly grasp the level of risk and the proposed actions and decisions. Reporting templates could be included in company’s standard operating procedures.
Communication can happen at all points of the process; it is not necessary to wait for a formalised report before updating stakeholders with results.
Communication includes:
Email.
Automated quality systems such as LIMS, SAP and other Change Control systems.
Reports.
Meetings.
Risk Review is the part of the risk management process where the output / report of the risk assessment is reviewed by all the stake holders. Any missing data, assumptions, knowledge gaps and uncertainty should be identified to all stakeholders, before agreed decisions are implemented. Any risk based decisions should be documented on implementation. In addition it may be necessary to re-assess those decisions once or if additional data, evidence or information comes to light since decisions taken where data or information is limited could be revised or further augmented.
Risk Assessment Tools:
Preliminary Hazard Analysis (PHA):
This approach is (in my opinion) mainly used to obtain a ‘ball park’ assessment of risk. It is usually a starting point for risk assessments or when limited information is available; it involves the following steps:
Risk identification.
A qualitative evaluation of hazard or harm.
Ranking of the risk in terms of a combination of severity of probability.
Identification of possible activities for mitigation or control.
Failure Mode Effects Analysis (FMEA):
This technique evaluates for potential failures. It could be applied to manufacturing process to assess the risk of failure of certain defined process steps and can be used to determine which process steps should have critical process parameters. This approach relies upon prior accumulated knowledge of the product and the process although information obtained from literature sources can also be utilised.
Fault Tree Analysis (FTA):
This is a process or method to establish or identify ‘root causes’ of risks by identifying all possible causes through a chain of events.
The purpose is:
To diagnose a problem
To understand and define possible root causes
To investigate the proposed root causes and
To propose mechanisms or actions to prevent, correct or mitigate them.
FTA provides a diagrammatic representation of the failure path or the failure chain of events and it involves a top down approach where all of the factors or components of a process are outlined and broken down into their essential components in order to establish the potential causes and to understand where risks can occur.
Risks could occur due to one factor (one root cause) or multiple factors (multiple root causes).
The FTA can also identify ‘undeveloped’ events which need to be further investigated as well as known ‘basic’ events. The root cause can be qualitative in nature as well as quantitative.
An understanding of the probability of each event is needed; the FTA can provide information on the dependencies of each event on the risk occurring. This can provide direction in terms of how to identify, monitor and control processes to prevent risks.
The FTA is useful not only for quality of products, but also for processes, e.g. regulatory submission in order to define regulatory strategy.
Whilst FTA is useful for identifying, assessing and controlling risk, it is also useful for issue resolution, particularly when problems have already occurred. It is a good problem solving tool.
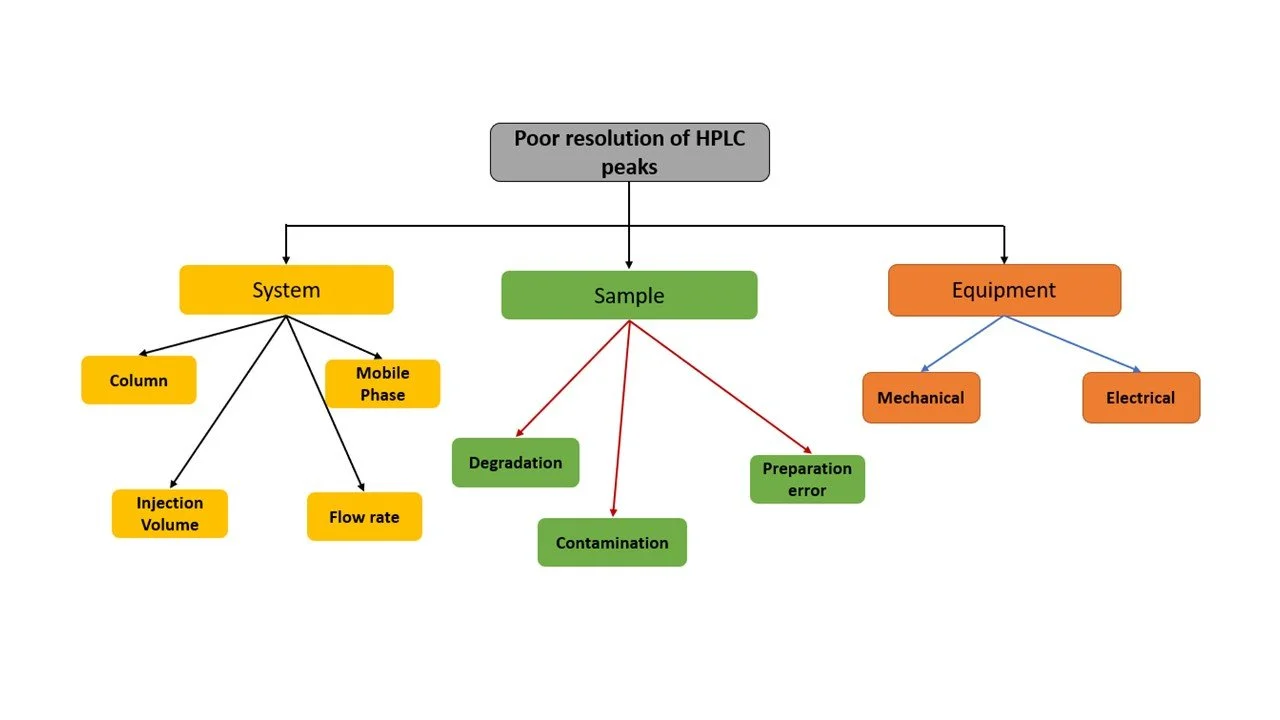
Example of a Fault tree analysis: Potential problems with new method: Poor resolution of HPLC peaks.
These can aid in defining the control parameters (e.g. the system parameters).
Fishbone Cause and Effect Analysis:
This is a tool that helps us to visualise potential factors (and causes) of a hazard, particularly if there are multiple factors and causes.
The hazard need to be correctly identified in the first instance and the potential factors or causes identified. These factors are the ‘categories’ of potential causes and are usually obtained through brainstorming. The brainstorming is usually the critical part of the fishbone cause and effect analysis.
An simple example of the fishbone cause and effect analysis is outlined in a diagram format as outlined below.
The information presented in diagram below is more easily analysed and allows actions (such as informed science based decisions) to be more apparent.
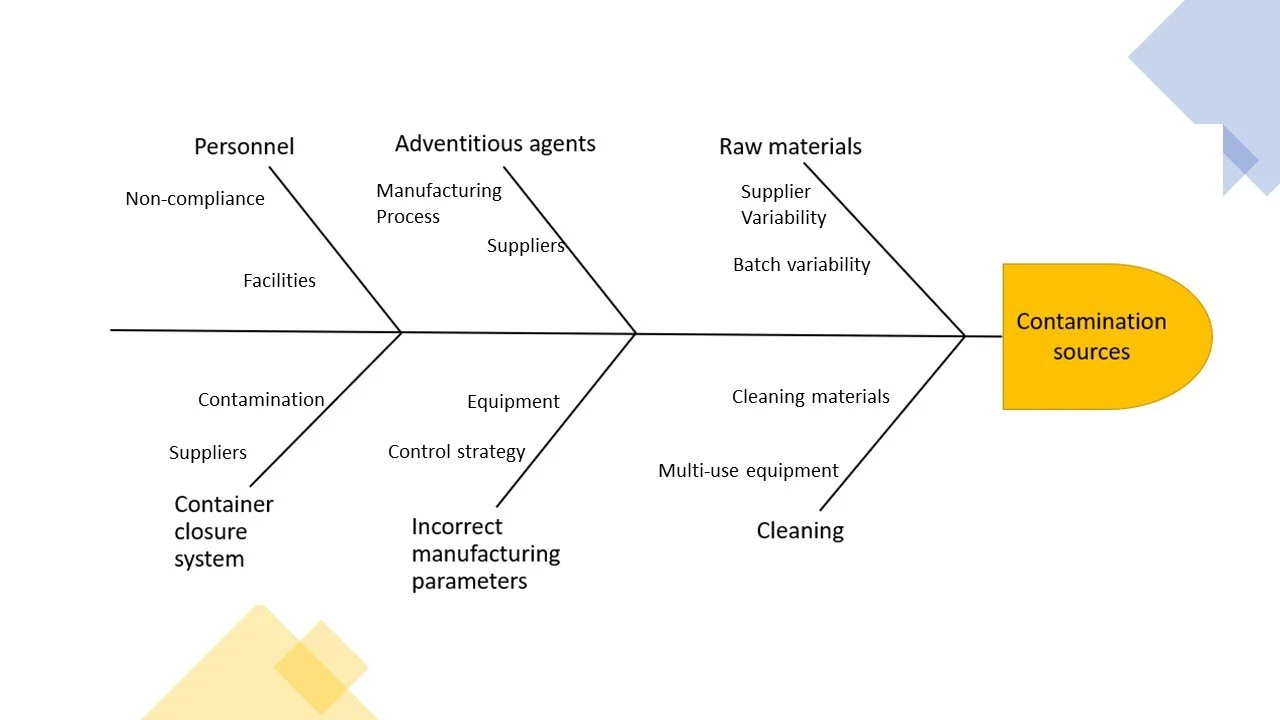
A fishbone cause and effect diagram can be used as a starting point for brainstorming.
Hazard Analysis and Critical Control Points (HACCP):
This is a systematic, proactive and preventive approach that applies scientific principles to analyses, evaluate, prevent and control risk of hazards and/or adverse consequences of hazard at specific control points (namely the critical control points). It can be a means to establish limits and critical process parameters in manufacturing processes as well as corrective measures.
Hazard and Operability Study
This process to identify risk of hazards through a creative and collaborative approach based on the collective knowledge of a multi-disciplinary team and a facilitator to ensure adherence to the process. For HAZOP studies it is important to be quite specific in relation to the subject or aspect under consideration - in other words a focussed approach.
The objective it to consider specific process deviations such as (high temperature, incorrect flow speed, incorrect mixing speed, etc.) and to evaluate worse case scenarios (e.g. failure) and the probability of occurence.
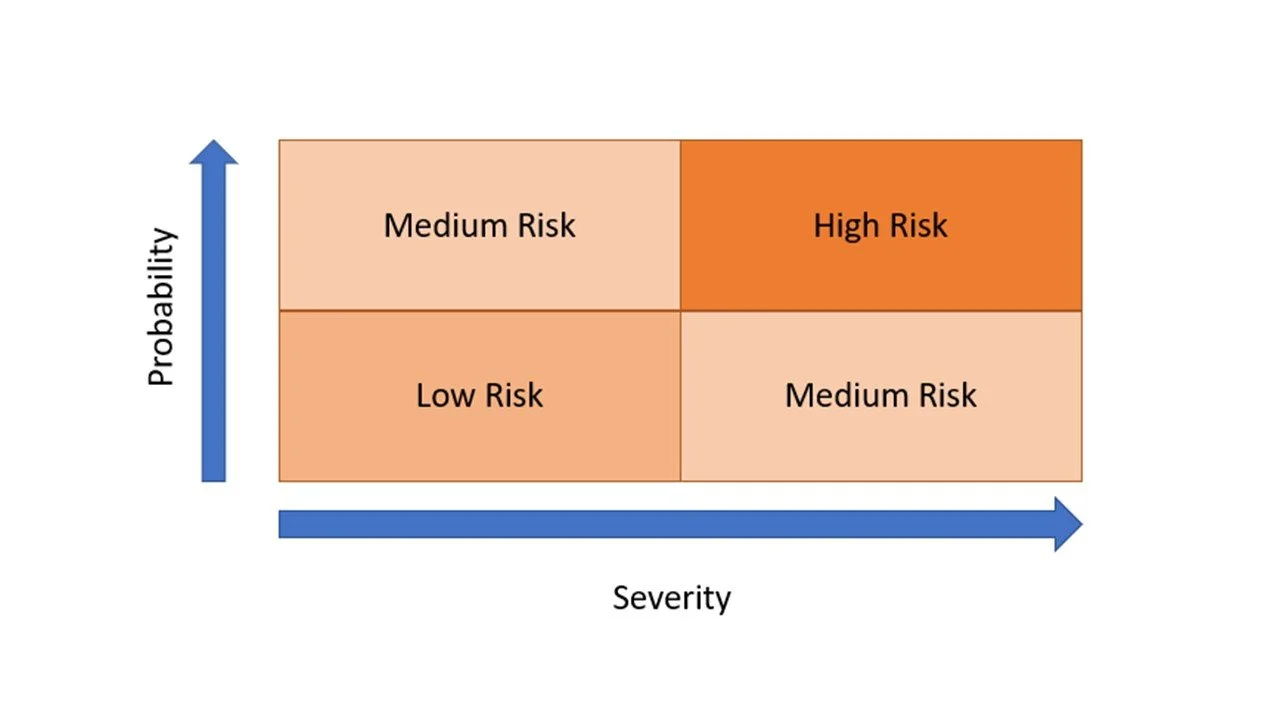
The level of risk from the identified hazard is evaluated by an assessment of the probability and severity with and without any safeguards and categorised according to the risk matrix.
This allows stakeholders to consider and agree upon risk based decisions and actions. Actions could be to accept the risk or lower the risk and take mitigation actions.
Risk Ranking and Filtering:
Risk ranking and filtering are a useful and approach and tools to utilise for multiple risks or hazards applied to a system, process (such as a manufacturing process change or issue) or procedure (such as a regulatory procedure). It is a process where each risk is ranked by qualitative (high, medium or low) or quantitative (percentage, discrete values etc.).
It is primarily used to prioritise risk in accordance with the level of severity and probability of hazard or harm occurring. It can be useful for simplifying priority for risks where multiple factors contribute to the risk by combining the risk score into a relative risk score.
Conclusion
Quality Risk Management is a useful approach in the pharmaceutical industry to improve efficiency by preventing quality defects and the subsequent recalls. The objective in the pharma industry is to be able to identify, control or mitigate hazards that impacts on patient safety.
In regulatory affairs, risk management tools can be used for project management activities including the use of risk registers to alert stakeholders of identified risks and to execute submission strategies.
Risk ranking is an effective tool to prioritise auditing and inspection activities as well as project management activities; risk ranking is also used for categorising changes in manufacturing processes when determining comparability.
Risk assessments are useful for preventative measures with respect to harm from hazards that may not be immediately apparent. Random incidents can occur; the objective of risk assessments is to minimise the impact of those occurrences by predictive assessments that enable pharma industry to take measures that will prevent loss of expensive medicinal product, defective medicinal products and/or product recalls due to quality issues.
As I go back to the ‘black swan’ theory - an absence of evidence is not evidence of absence. Risk assessments can therefore be used as science based predictive tools that lowers the harm caused by potential random events by the detection, mitigation and the implementation of control measures.
The SMARTER approach (Specific, Measurable, Assignable, Results, Timebound, Evaluation and Recording) could be applied for risk assessments.
It should also be noted that the performance of risk assessments does not obviate a company from making the required regulatory submissions. In some cases it is recommended that regulatory agencies’ advice is sought before the implementation of changes and sometimes before the assessment or protocol analysis of any proposed changes are performed in order to obtain regulatory agreement of the required data set.
I hope you enjoyed my blog, let me know your thoughts and any comments.
Best wishes,