Development and Validation of Analytical Procedures
Introduction
The aim of analytical methods is to qualify and/or quantify the active substance(s) and any other relevant substance(s) in the drug product in order to confirm the quality.
The development and validation of analytical methods occurs over the product lifecycle; the level of validation is dependent on the stage of development, the nature of the methodology and the use of the analytical method.
Later phase product development requires full validation of analytical methods demonstrating that they are fit for purpose. Analytical methods should be capable of being transferred from one laboratory to another; however that does depend upon the modality of the medicinal product(s); analytical method transfer can be challenging for certain cell and gene therapy medicinal products; this is due to inherent variability with analytical procedures for these medicines.
This blog provides an outline of my thoughts on the development of analytical procedures and the validation expectations from national authorities.
Analytical Procedures and Analytical Methods
Analytical procedures, as required for Clinical Trial Authorisations (CTA) and for Marketing Authorisation Applications (MAAs), are effectively summaries of analytical methods.
It is not good practice to include the full analytical method in an applications dossier since there are many reasons for updating analytical methods. Certain changes should be managed by the internal quality systems at the testing site in accordance with their Pharmaceutical Quality Systems (PQS).
It is not always necessary to include the level of detail required for the practical application of the analytical method in the laboratory, since that is not really what is being assessed; this falls under the scope of Good Laboratory Practice and not the submission documentation. In my experience, this is the approach taken in Europe, although I am aware that this varies in other international regions.
Analytical changes that impact on quality must be submitted to the national authorities via a modification/amendment for a CTA or variation application for a marketed product with a Marketing Authorisation (MA).
Information on analytical procedures for a MAA, will include but is not limited to, summaries of validation acceptance criteria, and validation data, sometimes with examples of the raw data including chromatograms. The analytical tests must be fit for purpose and at least qualified to a suitable level before first in human trials begin and certainly before applying for a MA.
Analytical Development
A risk assessment should always be conducted at the start of the product development for all aspects of the product (e.g. manufacturing, administration, dose, stability etc.). A more focussed risk assessment for the analytical method development for the active substance(s) and the final drug product should be performed by the analyst. The risk assessment is used to identify potential product and process related impurities, and possible degradation products.
Researching on similar medicinal products and the type of testing accepted and/or performed on them should also be prioritised and the information considered.
Analytical development begins with characterisation activities on the active substance. Information gleamed from this exercise will enable preliminary analytical methods to be identified. In addition, characterisation provides information on the preliminary critical quality attributes (CQA) that have a relationship to the clinical outcomes. Characterisation activities should also include an exercise to understand and define any potential microbial risk.
Analytical methods resulting from the characterisation activities should be defined, developed, and optimised to qualify and quantify proposed impurities and degradation products as well as the active substance.
During the product lifecycle, changes are going to be made on the manufacturing process and/or the product as a result of process improvements / optimisation or scale ups and scale out. Decisions made early in the development and validation of analytical methods should take this fact into consideration so that later comparability exercises can be provide more meaningful data that can be relied upon with confidence.
For the same reason, where possible, analytical methods that assess the stability of the active substance and drug product should also be considered early on in the product development. Analytical methods that are stability indicating could yield valuable information supporting or even accelerating the clinical development program.
There are obligatory CQAs that should be assessed as part of the testing strategy and those analytical methods should be developed, qualified and validated as soon as practicable.
Obligatory CQAs are tests that must be performed on the active substance(s) or drug product. They include identity, purity, potency, bioassay, sterility, endotoxin and physical characteristics of the active substance and the drug product.
As discussed above, the first step of analytical development in the laboratory is characterisation of the active substance(s) and/or the drug product. Characterisation is also performed on the manufacturing process. Characterisation tests that measure the impact of varying specific process parameters on CQAs, can be used to establish controls and monitoring parameters during production. An acceptance criterion for the CQA can be applied to the resulting In-Process-Control (IPC) test. Thus, analytical methods are not just applicable to active substance or drug product release.
Tests and acceptance criteria listed on specification section of the dossier are derived from characterisation exercises during development and batch data generated from manufacturing campaigns (such as demonstration batches, clinical trial batches, pilot batches, exhibit batches etc.).
Ideally testing should be designed to be completed within a few days to a week; however, some tests do have long lead times (e.g., microbiological test); the time taken to complete testing generally extends proportionally to the complexity of the active substance or medicinal product.
In summary, for analytical development, the best place to start is with a risk assessment; then research for similar medicinal modalities and the type of testing strategy employed for them and then perform a characterisation exercise. These should lead to a starting point for analytical methods which could then be further developed and optimised for the active substance, drug product or manufacturing process.
Pharmacopoeia provides a point of reference for some test methods expected by regulatory authorities. The Applicant should, as much as possible comply with pharmacopeial standards in the relevant regions; diverging from these standards approaches required good justification. This should be along the lines of, certain tests being irrelevant to the product concerned, or, improved testing procedures selected in place of the standard approach.
Analytical Methodology
Analytical methods should measure or identify the active substance, other related substances, and/or the drug product. The quality attributes being measured should as early as possible be related to the biological activity in vivo, the mechanism of action and/or the clinical outcomes.
All the better, if a relationship between the CQAs and the clinical outcomes is established as early as possible. The analytical methods are key to the development of the product.
As previously noted, analytical methods are also used to establish control parameters and conditions during the manufacture of the active substance(s) or drug product. A testing strategy should be established to enable control of critical and key process parameters during manufacture.
Identification
The identification test relies upon specific characteristics of the drug substance to distinguish them from related impurities or degradation products. This principle is applicable to all modalities. Identification is a requirement for all medicinal products; in my experience usually 1 independent test and one supporting test.
Regarding small molecules, identification test includes Fourier-Transform Infra-Red Spectrum (FT-IR), or mass spectrometry (MS).
Identification is achieved by obtaining results or raw data on a designated qualified standard and comparing subsequent standards and/or samples against this designated standard using identical conditions of analysis.
In addition, identification tests for small molecules and biologics can be integrated into other test methods such as purity/assay techniques for example, Liquid Chromatography-Mass Spectrometry (LC-MS).
Identification can be implied through specificity. Antibody binding to specific antigens during biological activity tests can be utilised for identification purposes.
Identification tests for different modalities are indicated in the tables below.
The nucleic acid of Gene Therapy Medicinal Products (GTMPs) has unique genetic profiles which can be defined and assigned as a standard; other standards and samples can be compared against this standard for identification.
Purity Assay
Typically, the assay method is measuring the quantity of the active substance(s). For small molecules, proteins and polypeptides and biologics, I have previously used LC-MS or affinity HPLC and rarely chemical titration techniques. The acceptance criteria for these assays are determined by considering the variability of the method and data obtained on previous batches and similar molecules. Validation of these methods should occur in line with the ICH Guideline validation of analytical procedures Q2.
Purity assays include those that measure the active substance(s), process and product related impurities, and degradation products.
For biological materials, my limited experience includes ELISA, bioassays, Isoelectric focusing (for purity determination), SDS-PAGE (for degradation products such as aggregation), and affinity and Size Exclusion Chromatography (SEC).
For cell based medicinal products, the measurement depends upon how the cells function and is product specific. The physical characteristics of cells (morphology) should be recorded; as should viability and their ability to perform their intended function, such analysis/measurements of secreted proteins or other molecules and the potential for differentiation and cell division.
Physical Tests
These are likely to be active substance and/or drug product specific. They include morphological, structural, viscosity, size, condition, and other functional aspects.
Other tests
Sterility, endotoxin, mycoplasma are examples of tests associated with the safety of the medicinal products. They are obligatory and the applicable pharmacopeial method and standards should be applied in accordance with the regional requirements.
I have listed some the types of testing in accordance with the modality in the tables below; the list is a snapshot and is not exhaustive.

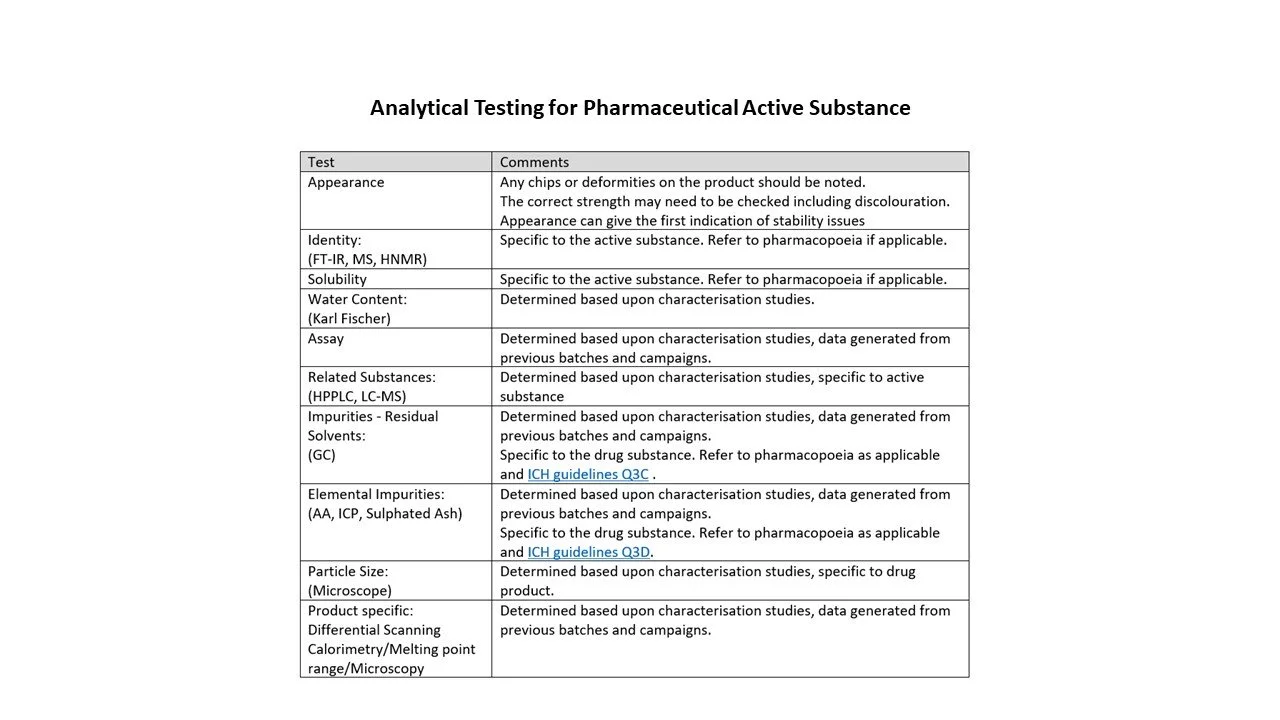

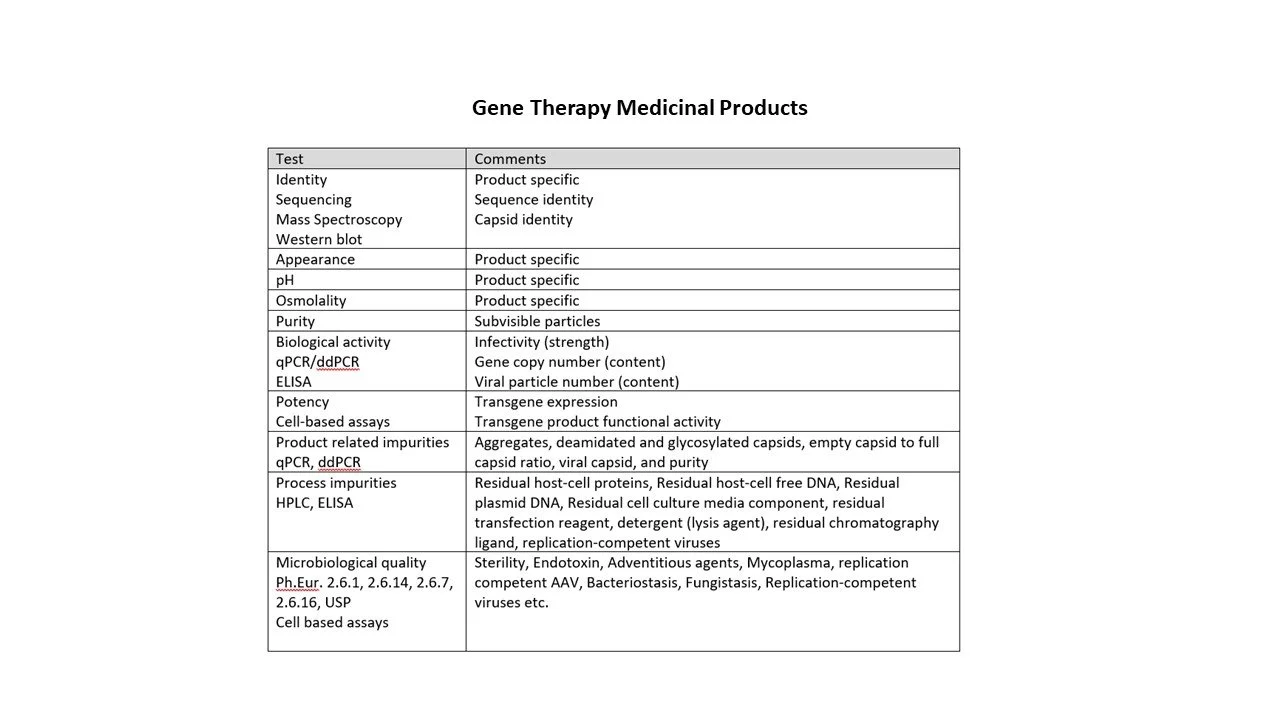
Analytical Validation
The validation of analytical methods demonstrates that they are fit for purpose and that the data generated from them can be relied upon.
During analytical validation, the acceptance criteria and the proposed method for statistical evaluation must be prospectively defined in a validation protocol before validation is initiated.
The parameters to determine are:
Specificity (active substance, impurities, and degradation products)
The specificity is the ability of the analytical procedure to distinguish and separate the active substances from other components in the sample (drug product sample). Specificity can also act as an identification test for the active substance(s), related substances and other excipients.
Linearity
The linearity of a range in an analytical method demonstrates that the response output is proportional to the amount of the substance that generates the output. Linearity is usually measured by correlation coefficient, y-intercept and the slope regression.
Range
The range of an analytical method describes the maximum and minimum limit that the method demonstrates linearity and is validated for.
Accuracy (active substance and impurities)
Accuracy is the ability of the method to provide a true and valid result. This is determined by applying known quantities of the material and determining the difference between the true result and the generated result.
Precision (repeatability, intermediate precision, reproducibility)
Precision measures the closeness of data from one another; it is separate to accuracy. One can have a number of inaccurate but precise measurements.
Repeatability looks at same system (equipment and reagents) on day one; intermediate precision looks at the different systems, different analysts on day two. Reproducibility looks at the method in a different laboratory with for example different manufacturer of the same equipment and different batches or suppliers of reagents.
Limit of Detection (LoD) and Limit of Quantification (LoQ)
The limits at which the active substance (or related substances) can be detected by the method. It looks at the signal to noise ratio of the method. The LoQ is the limits at which the active substance (or related substances) can be reliably quantified.
Robustness
Challenging the method to variability and measuring it’s performance by deliberately making small changes in certain parameters / conditions to see if the result is different from the results generated under the unchanged parameters / conditions.
System suitability testing
This is to determine that the analytical system (including the equipment and reagents) is functioning appropriately to yield the results as per the validated method. The system is challenged for precision, for accuracy and for ability to perform it’s desired critical function such as separating out components from one another on an HPLC column.
The ICH guidelines on the validation of analytical procedures Q2 should be adhered to and justification for deviating from these guidelines provided. Generally, analytical methods should be qualified for early development (such as nonclinical studies and clinical studies) and validated prior to marketing authorisation applications.
Replacing analytical methods with different methods must be carried out in a structured and controlled way; the new method must have data that demonstrates comparability between method and any data generated. The reasons for replacing analytical methods would be due to efficiencies in, time and improvement in accuracy and precision or other scientific rationales.
Analytical Verification
Verification is required for the implementation of pharmacopeial methods at a new testing sites. This can be system suitability tests or some other form of qualification. Verification are not full validation since pharmacopeial methods are already accepted as validated. They are just qualification tests to ensure that the testing site can perform the technique to the appropriate standard for the active substance or drug product.
In my experience, analytical verification can consist of system suitability tests to look at precision, accuracy and system performance.
Conclusion
Analytical development begins with a risk assessment, then literature researches around the active substance or similar active substances, and finally a characterisation exercise. Characterisation illuminates specific properties of the active substance that allows for the identification of an analytical methods for qualification, separation, and quantification of the active substance from other substances in the drug product (such as excipients and related substances). These methods can be further modified and optimised to measure the active substance in the final dosage form of the medicinal drug product.
Early in the development program, it is important to establish analytical methods that are at least qualified; this strengthens the evidence generated during comparability exercises later on in the product development.
Analytical methods should be established to support the testing strategy of the product, manufacturing process controls and other aspects, such as testing associated with cleaning validation.
Analytical validation should be conducted in line with the ICH guidelines.
I hope I have given a brief overview of analytical development and validation activities for the various medicines. Let me know if you have any further questions.
Best wishes…